
LA MASTOCITOSI INCLUDE UN INSIEME DI PATOLOGIE CLONALI RARE, CARATTERIZZATA DALLA PROLIFERAZIONE E DALL’ACCUMULO DI MASTOCITI PATOLOGICI A LIVELLO DI UNO O PIÙ TESSUTI, IN PARTICOLARE CUTE, MIDOLLO OSSEO, FEGATO, MILZA O TRATTO GASTROINTESTINALE.
Dal punto di vista clinico si manifestano in modo molto eterogeneo e spaziano da forme con clinica indolente e prognosi positiva, a forme aggressive con prognosi infausta.
Si distingue fondamentalmente una mastocitosi cutanea (cutaneous mastocytosis: CM), localizzata esclusivamente nella cute e tipica dell’età infantile, e una forma sistemica (Systemic mastocytosis: SM) caratteristica dei soggetti adulti, con coinvolgimento di almeno un organo extracutaneo, più frequentemente il midollo osseo, associato o meno a localizzazione cutanea. La sintomatologia è molto variegata, include sintomi da rilascio di mediatori mastocitari sia acuti che cronici, come flushing, prurito, anafilassi, disturbi gastrointestinali, osteoporosi/penia, fratture su base osteoporotica (più frequenti nelle forme indolenti), che sintomi da infiltrazione di mastociti che determinano danno d’organo con possibile epatomegalia, splenomegalia, linfadenopatia, anemia, leucopenia trombocitopenia e lisi ossee (caratteristici delle rare forme aggressive).
Dal punto di vista clinico si manifestano in modo molto eterogeneo e spaziano da forme con clinica indolente e prognosi positiva, a forme aggressive con prognosi infausta.
Si distingue fondamentalmente una mastocitosi cutanea (cutaneous mastocytosis: CM), localizzata esclusivamente nella cute e tipica dell’età infantile, e una forma sistemica (Systemic mastocytosis: SM) caratteristica dei soggetti adulti, con coinvolgimento di almeno un organo extracutaneo, più frequentemente il midollo osseo, associato o meno a localizzazione cutanea. La sintomatologia è molto variegata, include sintomi da rilascio di mediatori mastocitari sia acuti che cronici, come flushing, prurito, anafilassi, disturbi gastrointestinali, osteoporosi/penia, fratture su base osteoporotica (più frequenti nelle forme indolenti), che sintomi da infiltrazione di mastociti che determinano danno d’organo con possibile epatomegalia, splenomegalia, linfadenopatia, anemia, leucopenia trombocitopenia e lisi ossee (caratteristici delle rare forme aggressive).
Sulla base di dati del GISM la prevalenza dei disordini clonali mastocitari (clonal mastcell disorders: CMD) in toto e della SM in particolare nella popolazione adulta della regione Veneto erano di 11,2 e di 10,2/100.000 abitanti rispettivamente, simile a quanto riportato in letteratura. Nella provincia di Verona, dove si ritiene che la grande maggioranza dei casi sospetti, o già diagnosticati, siano inviati al gruppo, la prevalenza dei DCM e delle SM risultavano più elevati: 20,6 e di 17,2/100.000 abitanti, rispettivamente. Sempre nella provincia di Verona, l’incidenza media di nuovi casi di SM era 1,09/100.000 abitanti/anno.
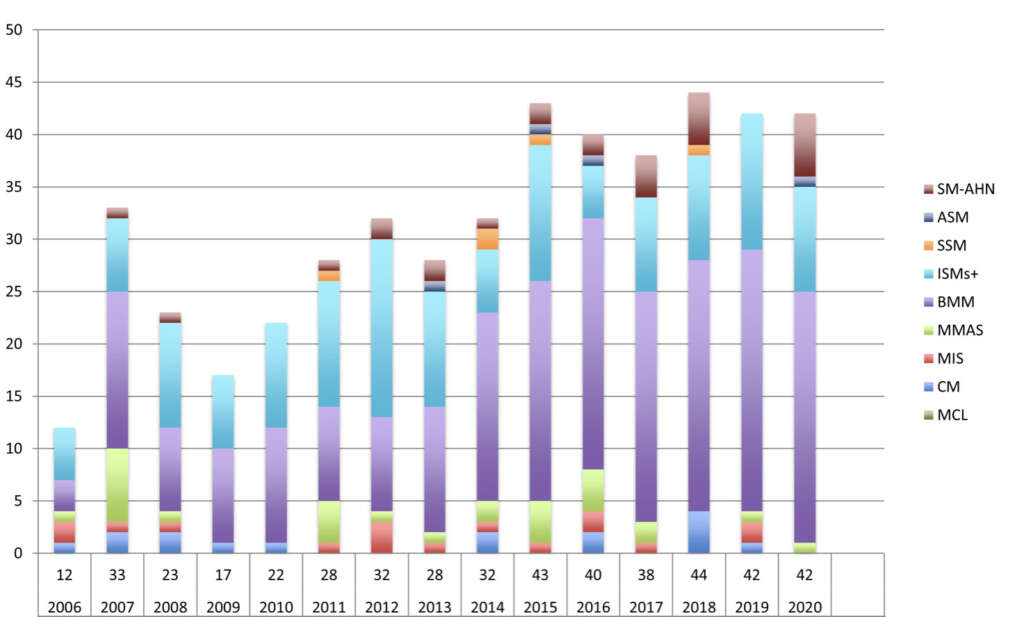
La forma di mastocitosi più frequente era l’indolente (ISM) (89%) ed in particolare la frequenza della sua variante limitata al midollo osseo (Bone marrow mastocytosis; BMM) (54,2%) era molto più elevata di quanto riportato in precedenza (10-39%). Nel 64% dei casi la BMM era diagnosticata solo sulla base di criteri minori WHO, che richiedono quindi l’utilizzo di tecniche diagnostiche sensibili con la citofluorimetria multiparametrica e la RT-PCR quantitativa per l’identificazione della mutazione D816V del gene KIT (tipica di circa il 90% delle mastocitosi sistemiche). L’allergia al veleno d’imenotteri (hymenoptera venom allergy; HVA) rappresentava il sintomo principale (50%) che portava alla diagnosi di CMD.
L’osteoporosi era presente nel 34% dei casi, spesso in soggetti giovani, soprattutto di sesso maschile. Circa un 60-70% dei pazienti con osteoporosi presentava già alla diagnosi fratture da fragilità, in genere vertebrali, spesso multiple (fino ad un massimo di 9 corpi vertebrali), e richiedeva terapia con bisfosfonati, per lo più per via endovenosa, oltre che supplemento con Vitamina D.
In conclusione, un approccio multidisciplinare è indispensabile per la corretta e precoce diagnosi di SM, ed in particolare delle BMM, spesso sottovalutate per l’assenza di coinvolgimento cutaneo. È inoltre importante che il paziente sia riferito in un centro specializzato con esperienza e tecniche diagnostiche sensibili per identificare i casi con scarso infiltrato mastcellulare. Una strategia di trattamento multidisciplinare è inoltre necessaria. La diagnosi precoce di CMD in pazienti con HVA, infatti, condiziona che l’immunoterapia specifica debba essere proseguita per tutta la vita, al fine di prevenire ulteriori reazioni allergiche severe, anche fatali. Il paziente con SM deve essere fornito di 2 autoiniettori di adrenalina per il trattamento di possibili eventi di anafilassi. Inoltre, l’identificazione precoce ed il trattamento dell’osteoporosi possono prevenire complicanze invalidanti a livello osseo.
Analogamente il centro di riferimento è necessario per il corretto e non semplice inquadramento diagnostico delle rare forme avanzate e il loro trattamento, oltre all’inserimento dei pazienti in protocolli sperimentali, data l’assenza ad oggi di una terapia in grado di guarire tale malattia.
Dott.ssa Roberta Zanotti